

O vírus da Gripe A (IAV) é um dos agentes patogénicos respiratórios mais importantes em suínos.
Tem um impacto sobre a mortalidade e causa perdas económicas significativas devido à descida de produtividade e os custos associados às vacinações e tratamento. Além disso, devido à susceptibilidade dos porcos a infecções com IAV de diferentes espécies, em concreto humanos, podem emergir novos vírus por reorganização genética que tenham implicações para a saúde pública. Em consequência, a compreensão da diversidade genética dos vírus circulantes em suínos, pode identificar novas linhagens virais e proporcionar um critério no qual basear e melhorar as estratégias de intervenção.

A diversidade genética do IAV suíno nos EUA é produto da deriva e alteração antigénica, adicionado ao facto da introdução de IAV de outras espécies nas populações suínas (Vincent, et al. 2008). Em termos gerais, há co-circulação de três subtipos dominantes (H1N1, H1N2 e H3N2) e quatro linhagens genéticas principais descritas segundo a origem dos segmentos génicos. A linhagem clássica suína H1 foi originada a partir da pandemia da gripe espanhola de 1918. A segunda linhagem foi detectada nos finais dos anos 90 a partir da hemaglutinina (HA) e da neuraminidase (NA) derivadas da gripe humana estacional H3N2. Esta linhagem era um vírus novo reordenado, que continha segmentos génicos HA, NA e PB1 derivados da gripe humana estacional H3N2, segmentos génicos PB2 e PA de Gripe Aviária e segmentos génicos NP, M e NS de Gripe Suína Clássica H1N1, pelo que foi denominado de vírus "reordenado triplo". Estes vírus reordenaram-se, por sua vez, com vírus clássicos H1N1 para adquirir a HA e/ou a NA, resultando em novos tipos genéticos de vírus H1N1 e H1N2. A constelação de genes internos da reordenação tripla (TRIG) permaneceu relativamente consistente com as origens suínas (genes M, NP e NS), aviárias (genes PB2 e PA) e humanos (PB1) dos vírus da gripe. A terceira linhagem produziu-se a partir de infecções repetidas do vírus humano estacional H1 IAV em porcos (o que é conhecido como "spillover", o evento no qual o agente patogénico próprio de uma espécie salta para outra espécie) no início do século XXI, criando duas linhagens diferentes de vírus humanos estacionais H1N1 e H1N2. A quarta, e mais recente, linhagem, representa um "spillover" do vírus humano estacional H3N2 que teve lugar em 2010-11. Dentro destas quatro linhagens principais de HA, o vírus continua a evoluir, formando novos ramos genéticos (figura 1).
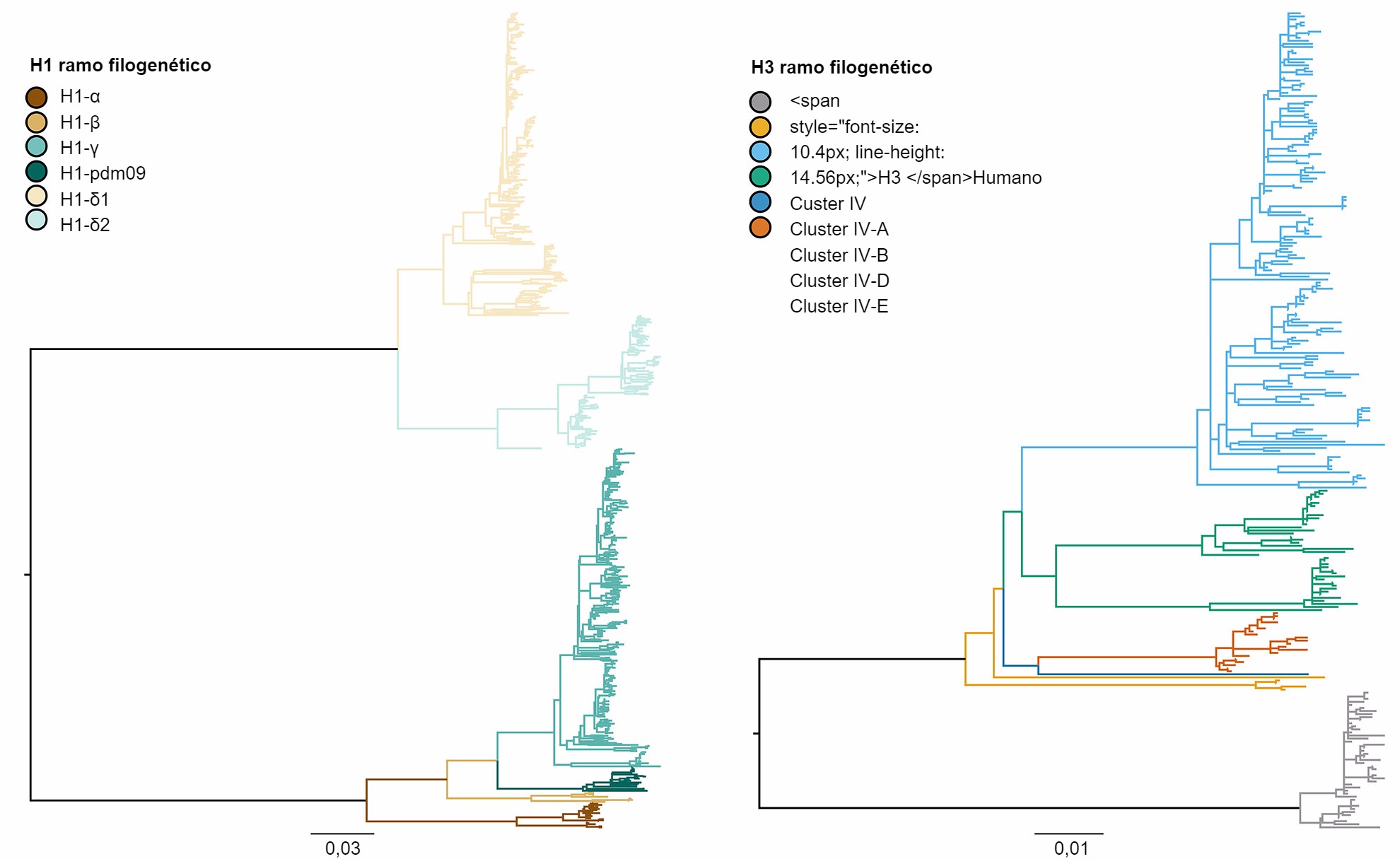
Figura 1. Árvore filogenética que descreve as relações genéticas entre sequências génicas da hemaglutinina da Gripe A, Suína H1 e H3 de 2015 geradas usando métodos de máxima verossemelhança. As ramificações em cor representam designações de ramo. As longitudes das ramificações estão desenhadas à escala e a barra de escala indica o número de substituições de nucleótidos por sitio.
Num esforço para melhorar a compreensão da diversidade genética do IAV suíno e monitorizar o vírus pandémico 2009 H1N1 (H1N1pdm09) em suíno, em 2009 foi colocado em funcionamento um sistema nacional de vigilância do departamento de agricultura dos EUA (USDA) que foi implementado através da rede nacional de laboratórios de sanidade animal (NAHLN). O sistema baseia-se em fornecimento anónimo de informações de produtores e veterinários, no entanto, o sistema padece de uma pobre informação geográfica sobre a procedência das amostras. Os dados epidemiológicos recolhidos incluem o estado, o tipo de amostra, o motivo de enviar, a idade, o tipo de localização, resultados da análise e, se aplicável, sequenciam-se os genes HA, NA e M e depositam-se na base de dados online de sequências do Centro Nacional de Informação Biotecnológica, GenBank (Korslund et al. 2012; Anderson et al. 2013). Estes dados, e o investimento contínuo no sistema, proporcionaram informação sobre os padrões de propagação do IAV, a sua diversidade genética ao longo do ano e a dinâmica da evolução do IAV na América do Norte desde 2010 até ao momento (Anderson et al., 2013; Anderson et al., 2015; Lewis et al. 2014; Rajão et al. 2015).
Os três subtipos (H1N1, H1N2 e H3N2) endémicos na população suína dos EUA foram detectados anualmente desde 2010 até à actualidade. Os subtipos H1N1 e H1N2 foram detectados com frequências semelhantes (~35%) durante os 5 anos. O subtipo H3N2 representa ~30% dos vírus identificados durante todo o periodo. Para descrever a diversidade genética dos vírus H1 é utilizado um sistema baseado nas letras do alfabeto grego para dividir os dados de ramos de HA dentro das linhagens clássicas (α, β, γ, γ-2 e H1N1pdm09) ou da linhagem humana (δ-1, δ-2). De um modo parecido, os genes do clúster IV de H3 são separados em 6 ramos genéticos (A a F) e um ramo H3 tipo humano (Rajão et al., 2015; Kitikoon et al. 2013). No entanto, pese ao potencial de circulação dos 14 ramos genéticos, a maioria da diversidade de HA observada nas explorações suínas dos EUA limita-se a três ramos. Desde Janeiro de 2015 a Dezembro de 2015, 43% dos isolados H1 pertenciam ao ramo γ da linhagem clássica e 37% foram classificados como δ-1 da linhagem humana estacional, os restantes 20% dos vírus H1 procederam dos ramos δ-2, α, H1N1pdm09 e β. Dos 8 ramos potenciais H3, o clúster IV-A representa a maioria de IAV H3 suínos circulantes nas explorações comerciais (61% dos vírus H3 durante 2015), foi identificado um número crescente de isolados como H3 tipo humano (de 5% em Dezembro de 2014 a 25% em Dezembro de 2015), enquanto que o resto dos isolados representam detecções esporádicas dos clústers IV-B, IV-C, IV-D, IV-E.
No contexto da eficácia dos programas de vacinação, há uma preocupação renovada de que a neuraminadase (NA) também possa desempenhar um papel importante devido à sua diversidade crescente (Sandbulte et al., 2016). Ainda que isto complique a situação, a NA tem muito menos linhagens que a HA. A HA está emparelhada com os genes N2 derivados de uma ou duas linhagens humanas estacionais H3N2 (sejam de 1998 ou de 2002: (Nelson et al., 2011)), genes N1 da linhagem clássica suína ou genes da linhagem estacional humana H1N1 (Anderson et al., 2013). Na actual IAV suína circulante, o N1 pertence, normalmente, à linhagem clássica (94%) e o N2 à linhagem 2002 (83%), enquanto que a linhagem 1998 representa, embora seja detectada de um modo consistente, um componente pequeno dos isolados IAV.
Uma segunda preocupação está relacionada com os padrões espaciais na diversidade de IAV suíno. Os EUA estão divididos em cinco regiões nas quais se reportam casos, baseando-se nos diferentes veterinários do USDA-APHIS (figura 2A). Há subtis e importantes diferenças entre regiões quanto à diversidade e abundância das amostras proporcionadas ao sistema de vigilância do USDA (figura 2B). Enquanto nas regiões 1 e 2 são detectados mais H1N1, a região 3 tem mais H1N2 e a região 4 mais H3N2. Embora o efectivo suíno seja de 1,6 milhões de porcos (USDA-NASS, 2012), há poucos dados da região 5. A região 2 mostra a maior diversidade em termos de diferentes ramos HA e NA observados e a maioria dos isolados de IAV suínos procedem desta região. Em geral, a distribuição de ramos HA e NA sugerem que as decisões regionais ou locais sobre os componentes das vacinas podem ser importantes.
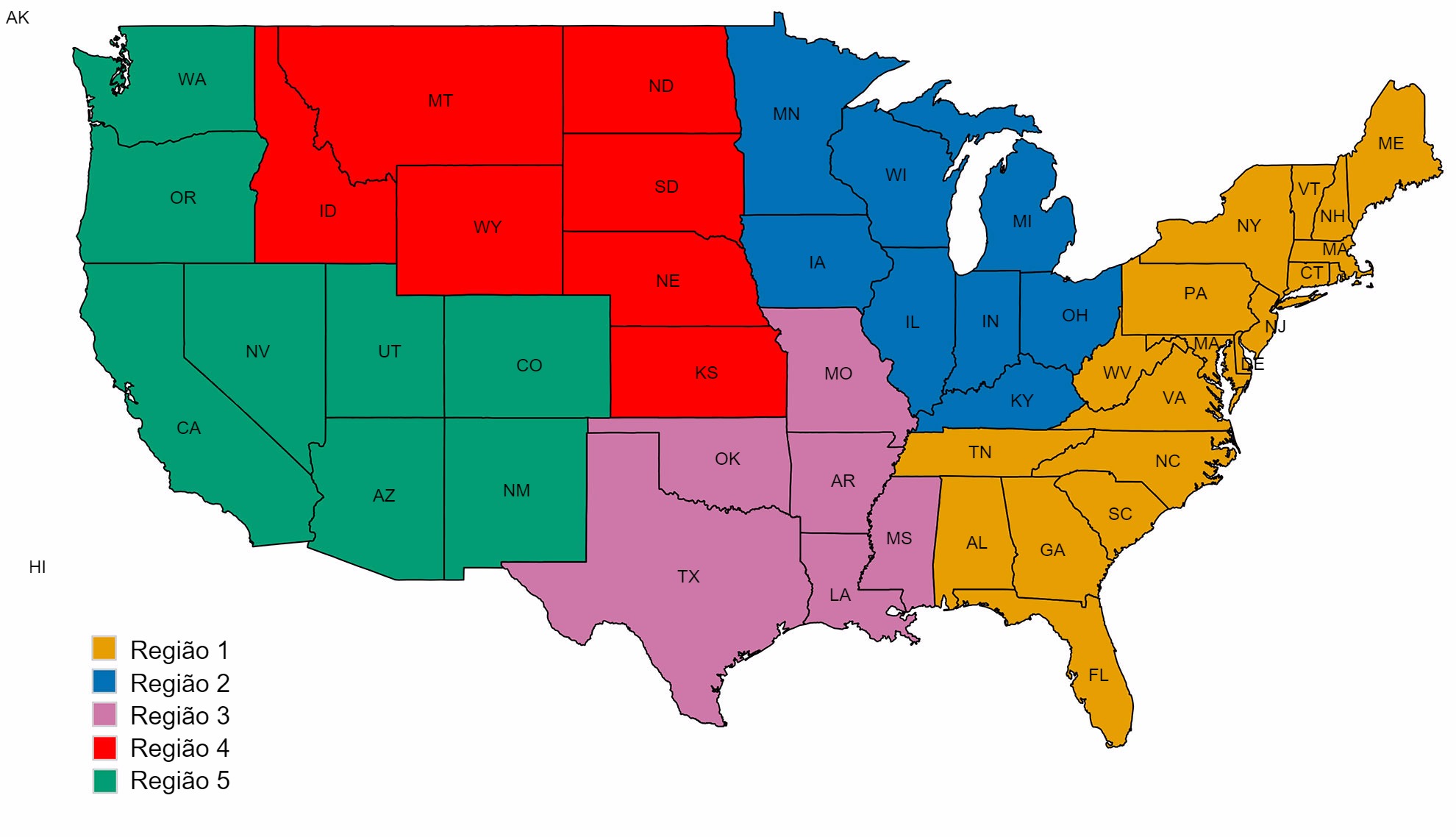
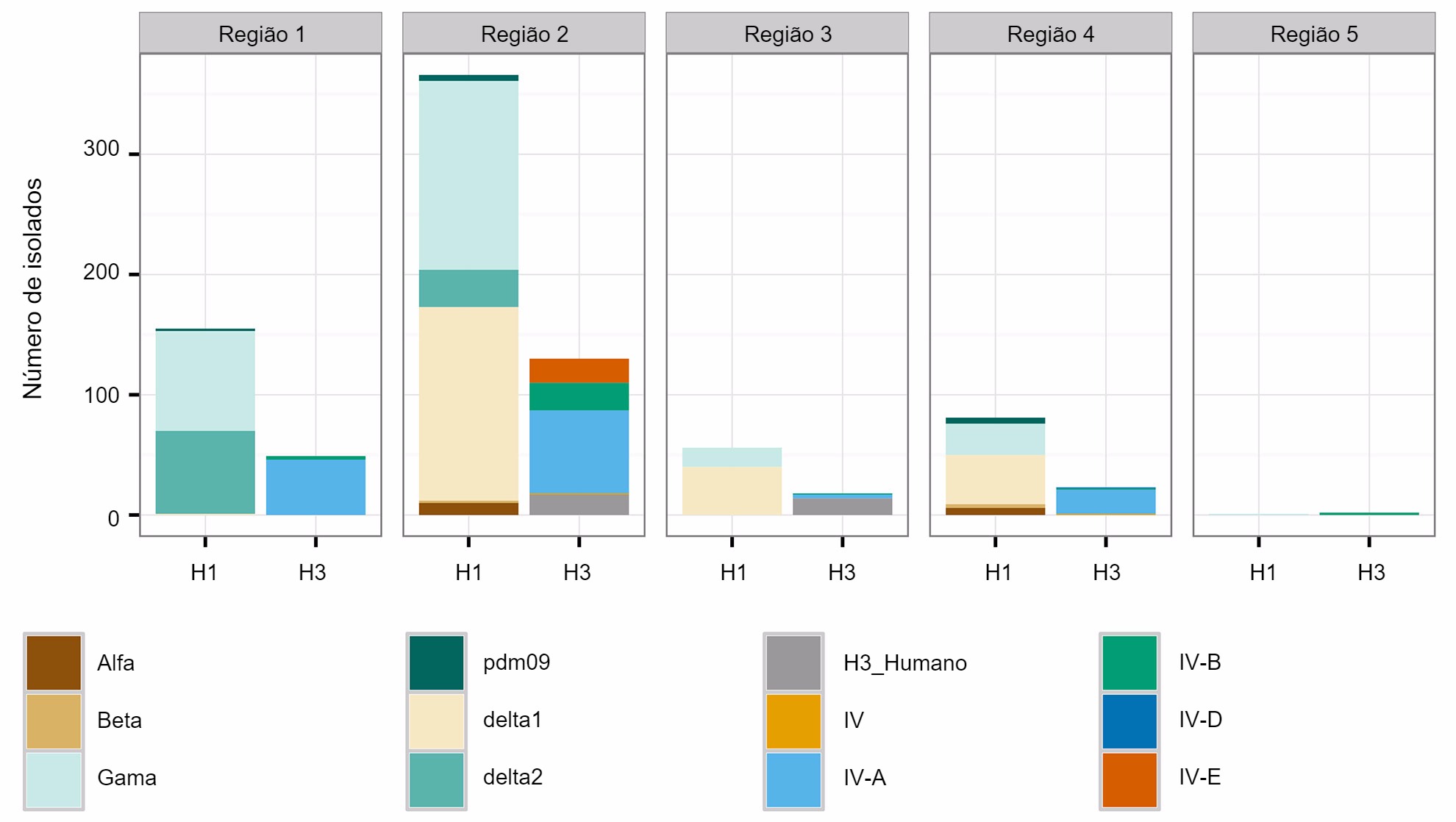
Figura 2. Regiões veterinárias do USDA-APHIS (A). Número de isolados de Gripe Suína A recolhidos em cada região durante 2015 e classificados segundo ramo filogenético e colorido como na figura 1 (B).
A diversidade genética do IAV suíno é um tema complexo a nível regional e, especialmente, a nível global. Nos EUA, houve 17 ramos genéticos que emergiram e persistiram a partir de eventos "spillover" entre hospedadores não suínos (ou seja, humanos) e os conseguintes processos ecológicos e evolutivos. Embora esteja constituída por diferentes ramos genéticos derivados de diferentes episódios de "spillover" não suínos, a diversidade genética é parecida na população suína mundial (por exemplo Watson et al. 2015; Bahl et al. 2015; Vijaykrishna D et al. 2015). A genética do IAV suíno contemporâneo pode ser utilizada para realizar estudos sobre evolução e diversidade antigénica e estes trabalhos deveriam ser realizados com dados actuais proporcionados por populações regionais. Dito conjunto de dados, com a implementação de uma plataforma vacinal apropriada, proporcionaria dados fundamentais para usar na selecção de estirpes vacinais e informação útil para as políticas de gestão de riscos para a sanidade animal e pública.

